Question: Rotation of diatomic molecules ([2] problem 3.6)
In our first look at the ideal gas we considered only the translational energy of the particles. But molecules can rotate, with kinetic energy. The rotation motion is quantized; and the energy levels of a diatomic molecule are of the form

where 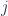


a
Find the partition function 

b
Evaluate 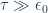
c
Do the same for 
d
Give expressions for the energy 

e
Sketch the behavior of 


Answer
a. Partition function
To understand the reference to multiplicity recall (section 4.13 [1]) that the rotational Hamiltonian was of the form

where the 
\begin{subequations}


\end{subequations}
and 



and our partition function is

We have no dependence on 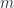

Fig 1: Summation over m
To get a feel for how many terms are significant in these sums, we refer to the plot of fig 2. We plot the partition function itself in, truncation at 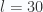

Fig 2: Plotting the partition function summand
 truncated after 30 terms in log plot")
Fig 3: Z_R(tau) truncated after 30 terms in log plot
b. Evaluate partition function for large temperatures
If 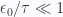

c. Evaluate partition function for small temperatures
When 


d. Energy and heat capacity
In the large 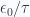

The specific heat in this domain is

For the small
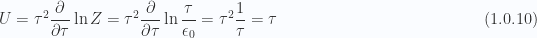
The heat capacity in this large temperature region is

which is unity as described in the problem.
e. Sketch
The energy and heat capacities are roughly sketched in fig 4.

Fig 4: Energy and heat capacity
It’s somewhat odd seeming that we have a zero point energy at zero temperature. Plotting the energy (truncating the sums to 30 terms) in fig 5, we don’t see such a zero point energy.
")
Fig 5: Exact plot of the energy for a range of temperatures (30 terms of the sums retained)
That plotted energy is as follows, computed without first dropping any terms of the partition function

To avoid the zero point energy, we have to use this and not the truncated partition function to do the integral approximation. Doing that calculation (which isn’t as convenient, so I cheated and used Mathematica). We obtain

This approximation, which has taken the sums to infinity, is plotted in fig 6.

Fig 6: Low temperature approximation of the energy
From eq. 1.0.12, we can take one more derivative to calculate the exact specific heat
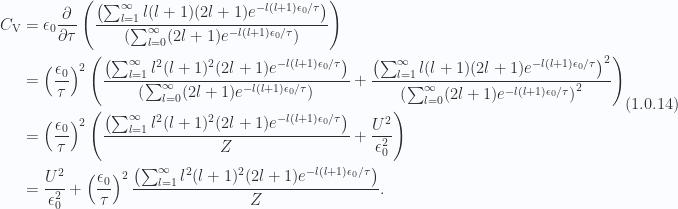
This is plotted to 30 terms in fig 7.

Fig 7: Specific heat to 30 terms
References
[1] BR Desai. Quantum mechanics with basic field theory. Cambridge University Press, 2009.
[2] C. Kittel and H. Kroemer. Thermal physics. WH Freeman, 1980.







 to express the number of states where the energy matches the average energy
to express the number of states where the energy matches the average energy  .
.

 as
as





 and
and  for the states
for the states  and
and  respectively, is given by the sum
respectively, is given by the sum
 are many body energies, so that
are many body energies, so that  .
.

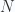 spin
spin  objects
objects  ,
,  , satisfying
, satisfying
 we have
we have
 where
where  . The total number of states is
. The total number of states is  .
.










 . This is the thermodynamic entropy introduced by Boltzmann (microscopic).
. This is the thermodynamic entropy introduced by Boltzmann (microscopic).







 and
and  , the exponential can be approximated by
, the exponential can be approximated by




 and the reservoir
and the reservoir  so that with
so that with







 the average energy is
the average energy is
















 from the constraint specified in the problem, but I’m guessing that constant factors of that sort have just been dropped.
from the constraint specified in the problem, but I’m guessing that constant factors of that sort have just been dropped. . With
. With  being an operator in the QM context, what did this even mean. I found the answer in [1] section 12.12. Here
being an operator in the QM context, what did this even mean. I found the answer in [1] section 12.12. Here 
 is given by eq. 1.0.4a. The rest of the problem can also be found there and relies on the WKB connection formulas, which aren’t derived in any text that I own. Quoting results based on other results that I don’t know the origin of it’s worthwhile, so that’s as far as I’ll attempt this question (but do plan to eventually look up and understand those WKB connection formulas, and then see how they can be applied in a problem like this).
is given by eq. 1.0.4a. The rest of the problem can also be found there and relies on the WKB connection formulas, which aren’t derived in any text that I own. Quoting results based on other results that I don’t know the origin of it’s worthwhile, so that’s as far as I’ll attempt this question (but do plan to eventually look up and understand those WKB connection formulas, and then see how they can be applied in a problem like this). included within a phase space trajectory. With coordinates as in fig. 1.1, our Lagrangian is
included within a phase space trajectory. With coordinates as in fig. 1.1, our Lagrangian is

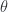 from the Euler-Lagrange equations
from the Euler-Lagrange equations  as expected. For the Hamiltonian, we need the canonical momentum
as expected. For the Hamiltonian, we need the canonical momentum
 ).
).

 , it is convient to non-dimensionalize this
, it is convient to non-dimensionalize this














 , is also this area. This is a general property, which can be seen geometrically in fig. 1.3, where we see that the counterclockwise oriented integral of
, is also this area. This is a general property, which can be seen geometrically in fig. 1.3, where we see that the counterclockwise oriented integral of  would give the negative area. The integrals along the
would give the negative area. The integrals along the  paths give the area under the blob, whereas the integrals along the other paths where the sense is opposite, give the complete area under the top boundary. Since they are oppositely sensed, adding them gives just the area of the blob.
paths give the area under the blob, whereas the integrals along the other paths where the sense is opposite, give the complete area under the top boundary. Since they are oppositely sensed, adding them gives just the area of the blob.




 ) and heating and cooling processes (with
) and heating and cooling processes (with  ).
).
 , a cyclic change is that for which we have
, a cyclic change is that for which we have





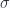 , as yet undefined. We call
, as yet undefined. We call 






 . Should we do this, we effectively also define
. Should we do this, we effectively also define 
 back to the initial values. This is characterized by
back to the initial values. This is characterized by



 follows from our definition
follows from our definition




















 ). We can think about an
). We can think about an  dimensional space, where
dimensional space, where






 , however, this does not imply
, however, this does not imply 






























 result for the thermal average energy.
result for the thermal average energy.
 as above. This gives us
as above. This gives us
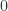 and one at energy
and one at energy  . From the free energy, find expressions for the energy and entropy of the system.
. From the free energy, find expressions for the energy and entropy of the system.





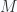 and the susceptibility
and the susceptibility  as a function of temperature and magnetic field for the model system of magnetic moments in a magnetic field. The result for the magnetization, found by other means, was
as a function of temperature and magnetic field for the model system of magnetic moments in a magnetic field. The result for the magnetization, found by other means, was  , where
, where  is the particle concentration. Find the free energy and express the result as a function only of
is the particle concentration. Find the free energy and express the result as a function only of  . Show that the susceptibility is
. Show that the susceptibility is  in the limit
in the limit  .
.




 , the cosh term goes to unity, so we have
, the cosh term goes to unity, so we have
 , the free energy is
, the free energy is
 , where
, where  is a positive integer or zero, and
is a positive integer or zero, and 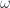 is the classical frequency of the oscillator. We have chosen the zero of energy at the state
is the classical frequency of the oscillator. We have chosen the zero of energy at the state  . Show that for a harmonic oscillator the free energy is
. Show that for a harmonic oscillator the free energy is
 we may expand the argument of the logarithm to obtain
we may expand the argument of the logarithm to obtain  . From 1.0.16 show that the entropy is
. From 1.0.16 show that the entropy is
 from the energy. Including it we have
from the energy. Including it we have


 in the energy of each state just adds a constant factor to the free energy. This will drop out when we compute the entropy. Dropping that factor now that we know why it doesn’t contribute, we can complete the summation, so have, by inspection
in the energy of each state just adds a constant factor to the free energy. This will drop out when we compute the entropy. Dropping that factor now that we know why it doesn’t contribute, we can complete the summation, so have, by inspection


 . Hint: Use the partition function
. Hint: Use the partition function  to the mean square fluctuation. Also, multiply out the term
to the mean square fluctuation. Also, multiply out the term  .
.




